
Department of Physics and Engineering Physics,
Tulane University
"The SCAN density functional, its performance on open-shell d- and f-electron compounds revealing DFT error origins, and a solution to its numerical problem"
Feb 24, 2021 Schedule:
- Virtual Tea Time
- 03:00 to 03:30 PM Eastern Time (US and Canada)
- Virtual Colloquium
- 03:30 to 04:30 PM Eastern Time (US and Canada)
Abstract:
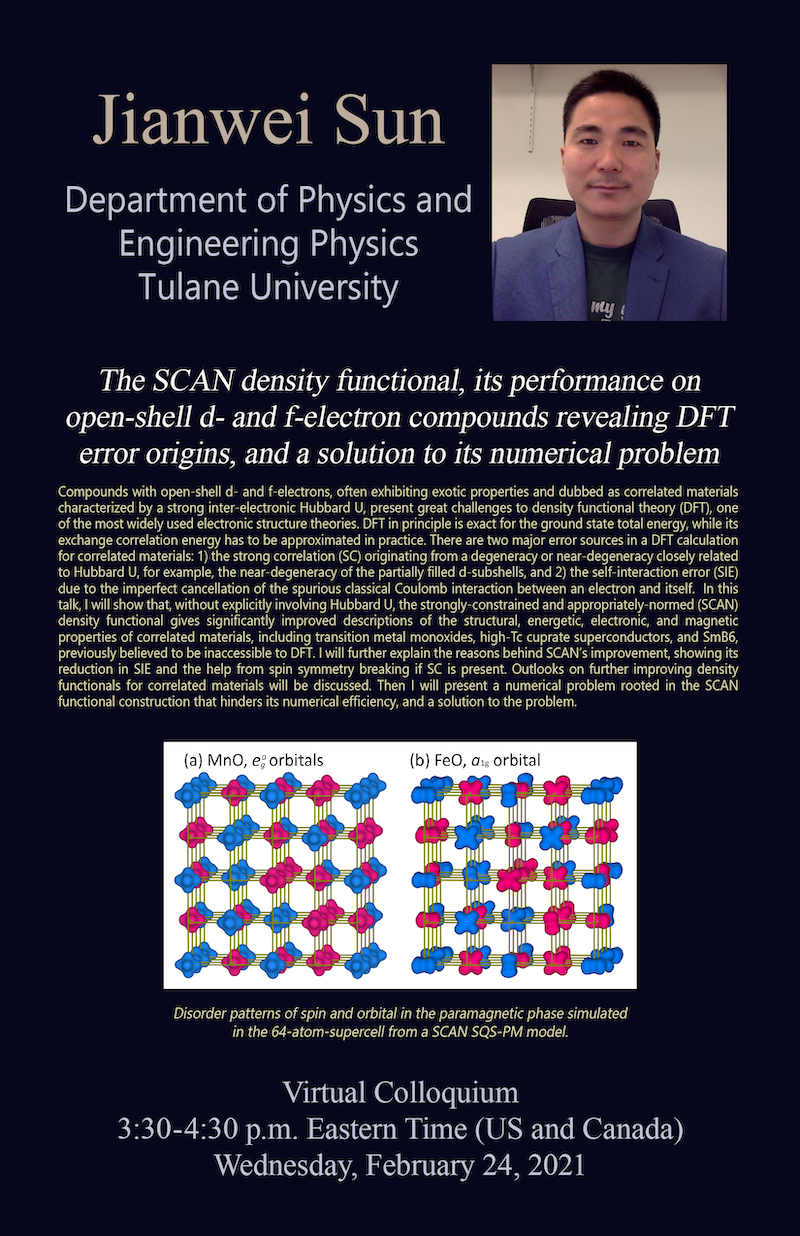
Compounds with open-shell d- and f-electrons, often exhibiting exotic properties and dubbed as correlated materials characterized by a strong inter-electronic Hubbard U, present great challenges to density functional theory (DFT), one of the most widely used electronic structure theories. DFT in principle is exact for the ground state total energy, while its exchange correlation energy has to be approximated in practice. There are two major error sources in a DFT calculation for correlated materials: 1) the strong correlation (SC) originating from a degeneracy or near-degeneracy closely related to Hubbard U, for example, the near-degeneracy of the partially filled d-subshells, and 2) the self-interaction error (SIE) due to the imperfect cancellation of the spurious classical Coulomb interaction between an electron and itself. In this talk, I will show that, without explicitly involving Hubbard U, the strongly-constrained and appropriately-normed (SCAN) density functional gives significantly improved descriptions of the structural, energetic, electronic, and magnetic properties of correlated materials, including transition metal monoxides, high-Tc cuprate superconductors, and SmB6, previously believed to be inaccessible to DFT. I will further explain the reasons behind SCAN’s improvement, showing its reduction in SIE and the help from spin symmetry breaking if SC is present. Outlooks on further improving density functionals for correlated materials will be discussed. Then I will present a numerical problem rooted in the SCAN functional construction that hinders its numerical efficiency, and a solution to the problem.
| File | Description | File size |
|---|---|---|
 2021-02-24sun.jpg 2021-02-24sun.jpg | Advertisement | 989 kB |
